Article’s abstract
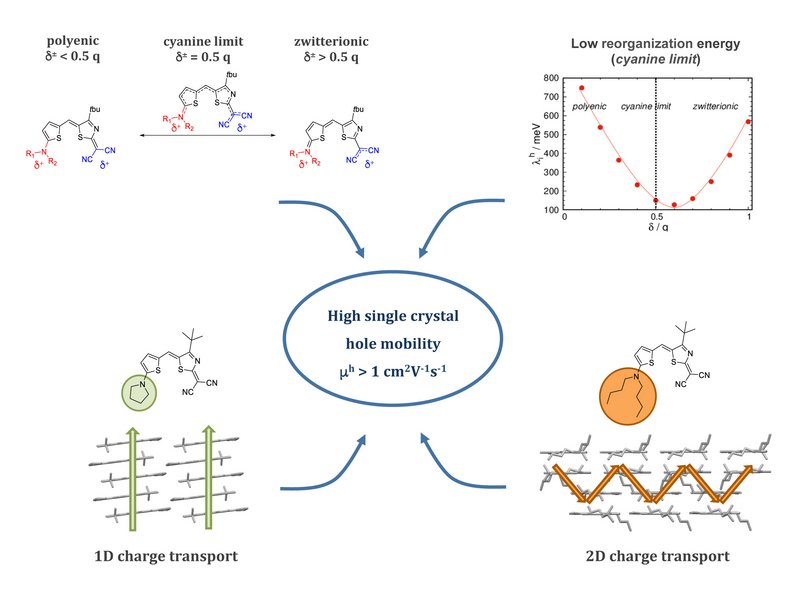
Merocyanines consist of electronic donor (D) and acceptor (A) subunits connected via a methine bridge. They are highly polar organic π-conjugated molecules investigated for their self-assembly and optoelectronic properties. The accurate description of their structure–property relationships remains challenging. We report a comprehensive analysis modelling intra- and inter-molecular charge transport parameters for a library of merocyanines featuring different D/A combinations and lateral substituents. We found that constrained DFT correctly assesses the molecular and electronic structure in single crystals. The most effective charge transport pathways were identified and charge carrier mobilities were computed. We analyzed a large variety of single crystals highlighting the impact of alkyl substituents and casting conditions, drawing clear structure vs. charge transport relationships. Our modelling suggests that hole transport is maximized when dipolar molecules are packed in slipped not centrosymmetric pairs, arranged in 2D interconnected architectures. Computed and experimental charge mobilities for single crystals are in good agreement.
Read the full article here: Link